PBDMine-Data¶
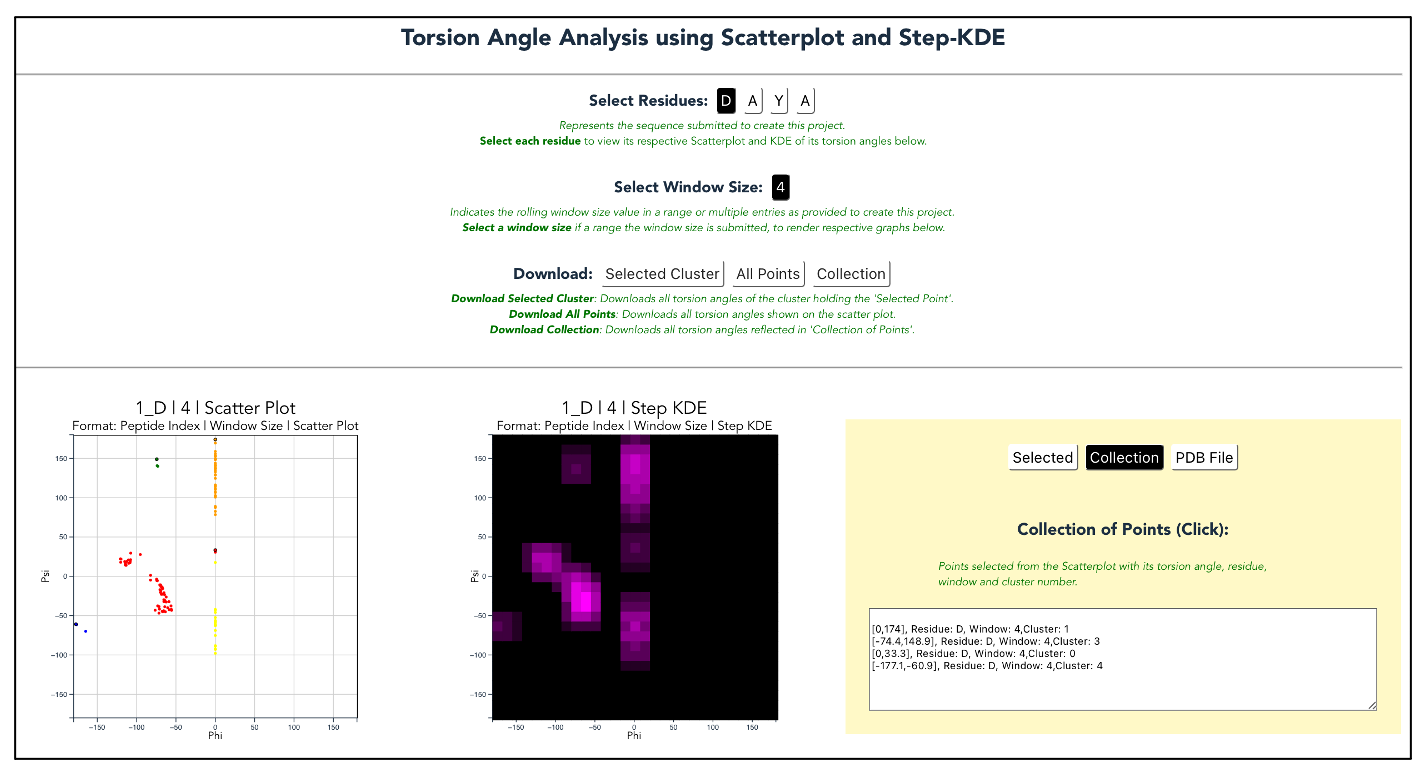
PDBMine discovers existing protein structures with a similar k-mer sequence of the protein; this structural similarity helps construct its fragmented structure. PDBMine returns torsion angles, which our website clusters and displays in a scatter plot. Furthermore, PDBMine - Data can use these angles to generate a pdb file modeling the potential structure of the protein.
A web application built with Python, Django, Vue.js.
Description¶
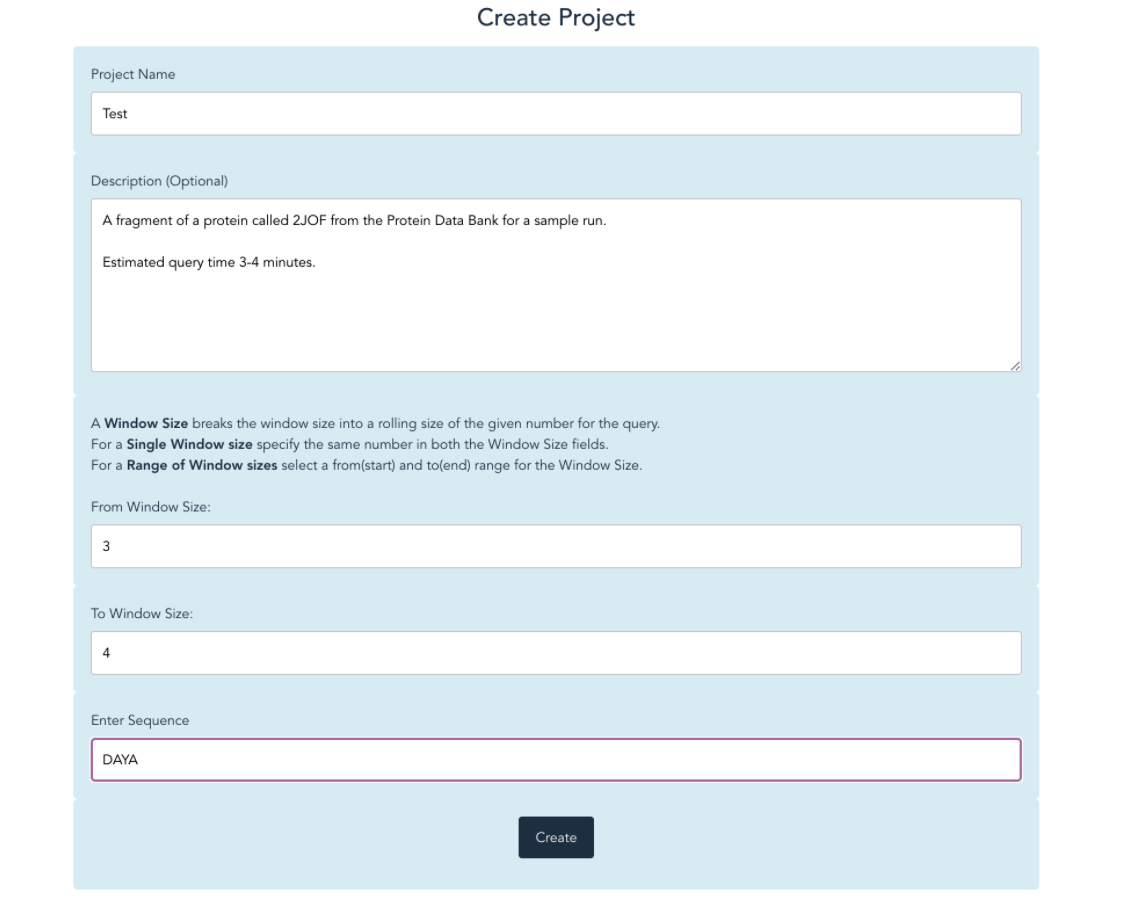
Our objective is to provide efficient computational solutions to better analyze genomic data, i.e., protein structure predictions based on torsion angles (phi-φ and psi-ψ)of previously classified proteins from the Protein Data Bank (PDB) to model a given/unknown amino acid sequence considering a rolling window size (k-mer). K-mer, where k is a given number of amino acid fragments of the user’s interest.
Authors¶
Claire Sturgill, Jenna Pehowski, Timothy Hooks, Naga Venkata Sai Jagjit Satti, Niharika Pandala.
Client¶
Dr. Homay Valafar, CSE Dept.
Screenshots¶